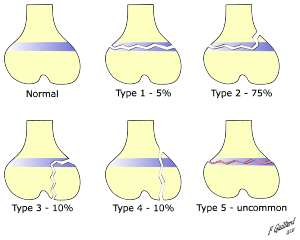
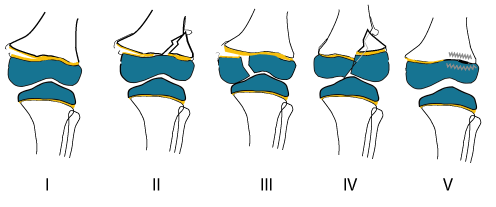
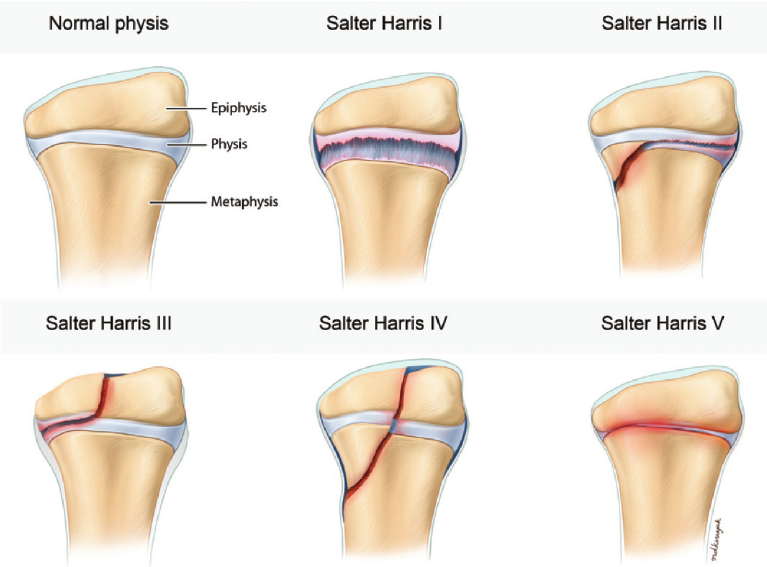
Salter-Harris fracture
“A Salter-Harris fracture is an injury to the growth plate area of a child’s bone.
The growth plate is a soft area of cartilage at the ends of long bones. These are bones that are longer than they are wide. Salter-Harris fractures can occur in any long bone, from fingers and toes, to arm and leg bones.
A child’s bone growth occurs mainly in the growth plates. When children are fully grown, these areas harden into solid bone.
The growth plates are relatively weak and can be injured by a fall, a collision, or excessive pressure. Salter-Harris fractures make up 15 to 30 percentTrusted Source of bone injuries in children. Most commonly these fractures occur in children and teenagers during sports activity. Boys are twice as likely as girls to have a Salter-Harris fracture.
It’s important to diagnose and treat a Salter-Harris fracture as soon as possible to ensure normal bone growth.”
Pandemic palliative care: beyond ventilators and saving lives
-
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic will likely strain our health care system beyond capacity, and palliative care services will be needed across many different care settings, including intensive care units, hospital wards, emergency departments and long-term care.
-
Shared decision-making between clinicians and patients is a core process in planning for the end of life; however, in a pandemic, patient autonomy to choose life-prolonging measures or location of death could be severely restricted as a result of public health directives and resource availability, and some patients may necessarily be isolated at end of life.
-
Previous mass casualty events have taught us much about how best to triage patients requiring care, and some of this work can be adapted to palliative care; but little has been written on how to manage those who are not offered life-sustaining measures.
-
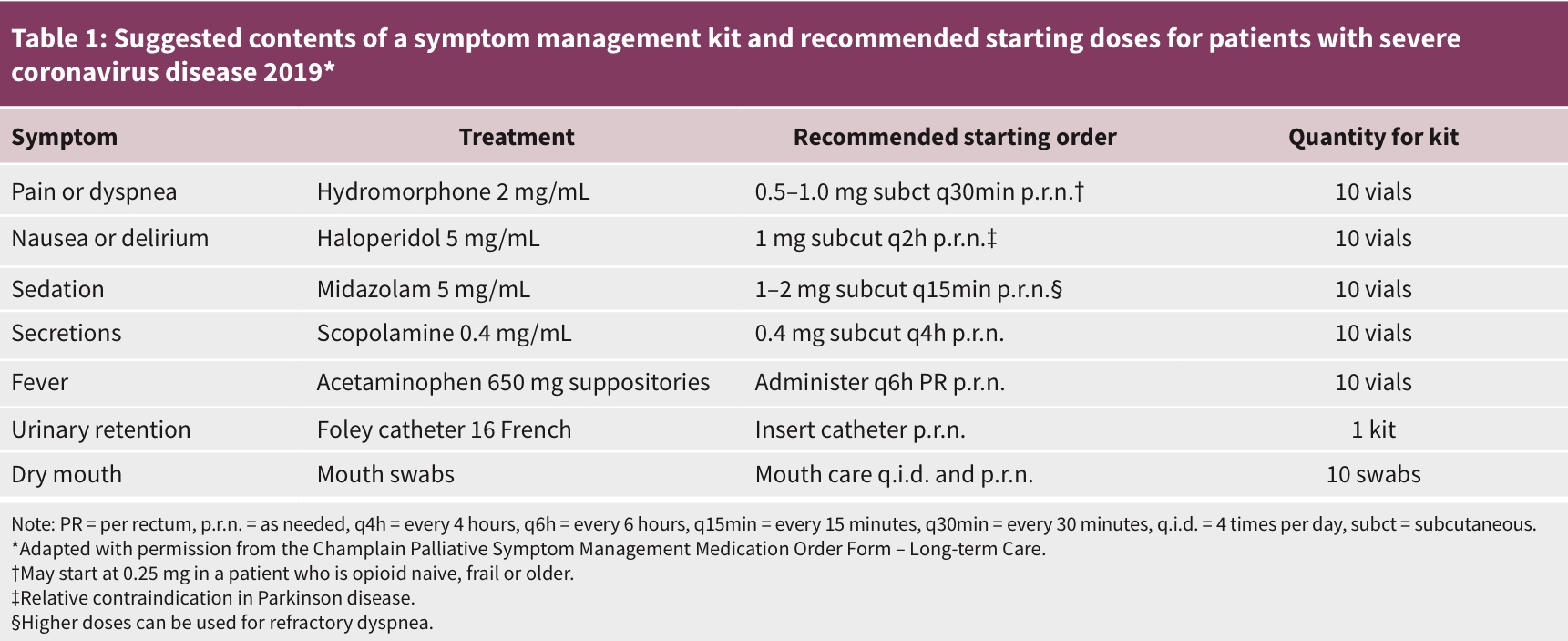
We advise acting now to stockpile medications and supplies used in palliative care, train staff to meet palliative care needs, optimize our space, refine our systems, alleviate the effects of separation, have critical conversations and focus on marginalized populations to ensure that all patients are cared for equitably.
-
The SARS-CoV-2 pandemic has been tragic for many people worldwide. Failing to provide Canadians with effective palliative care would compound that tragedy.
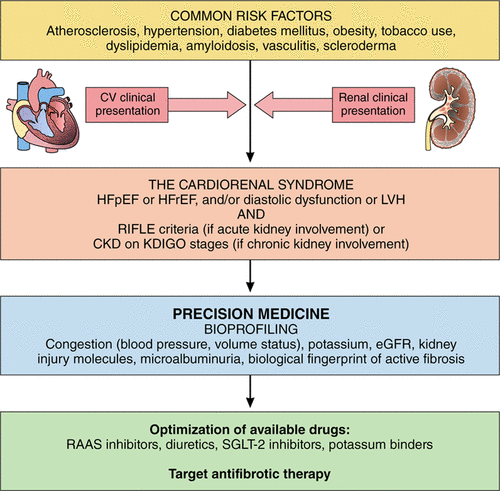
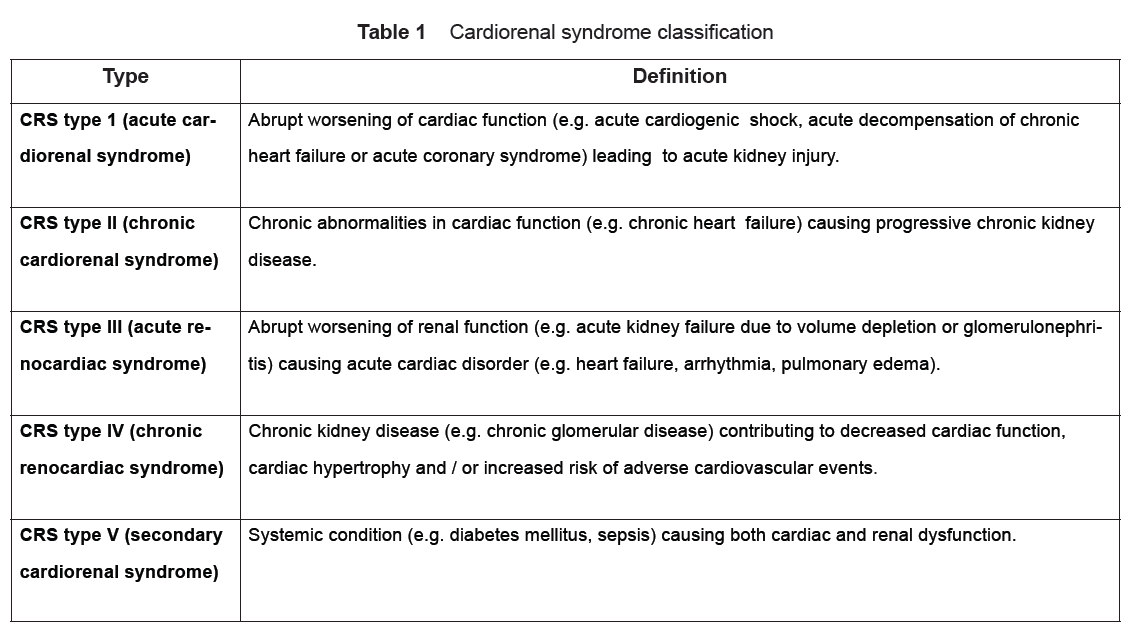
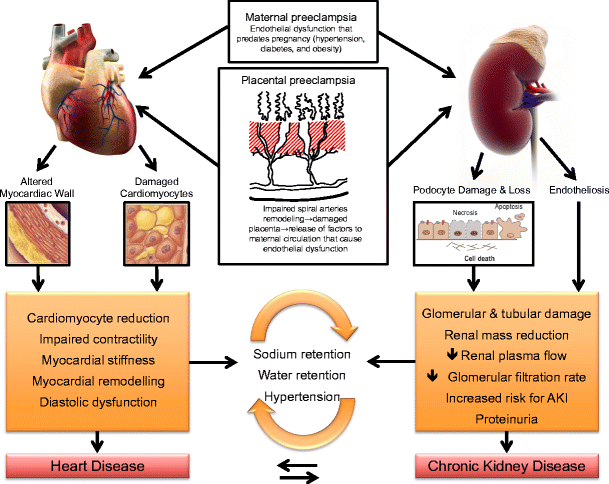
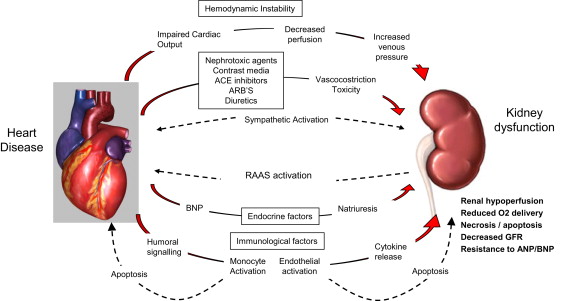
Cardiorenal syndrome (CRS)
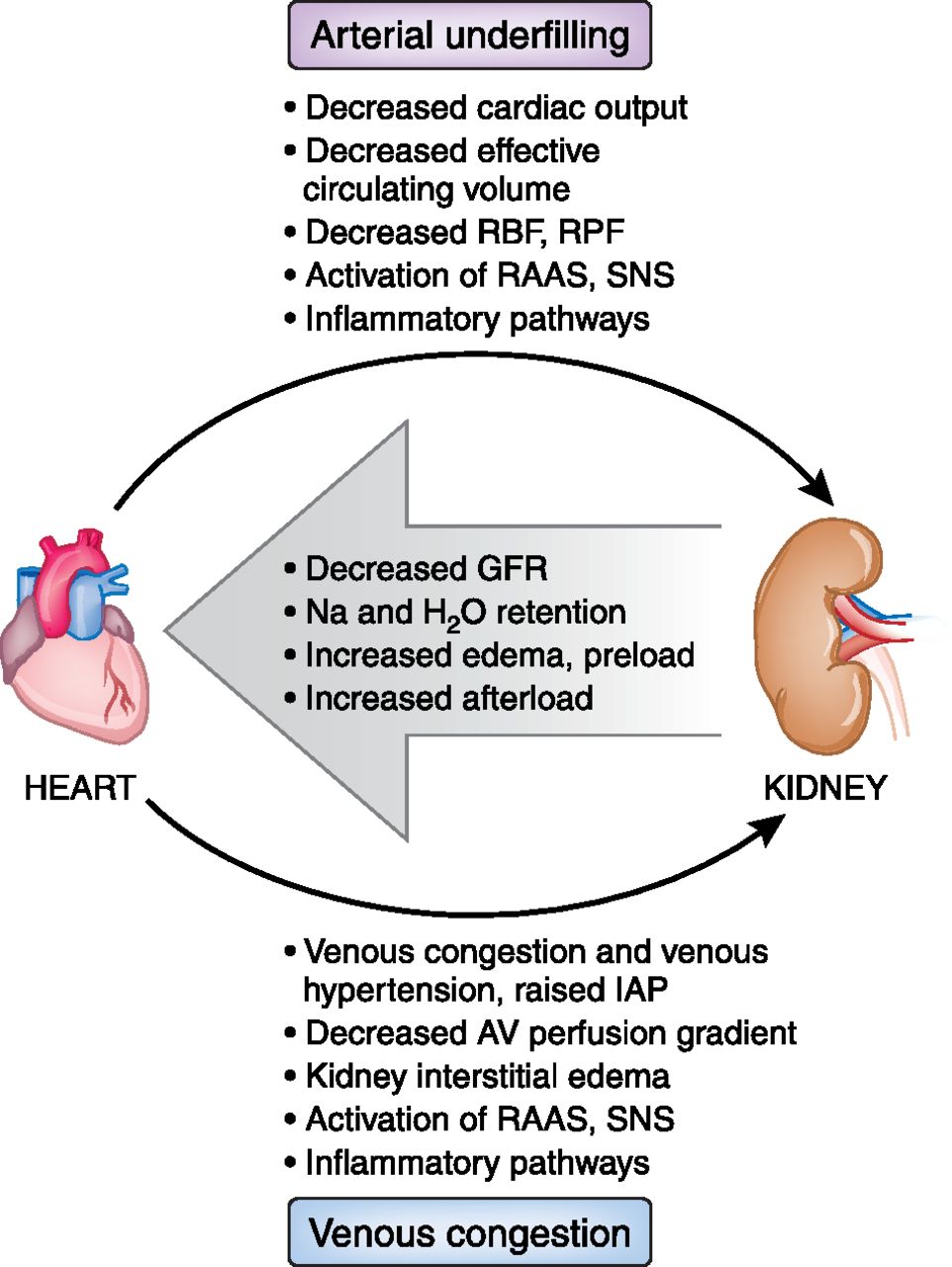
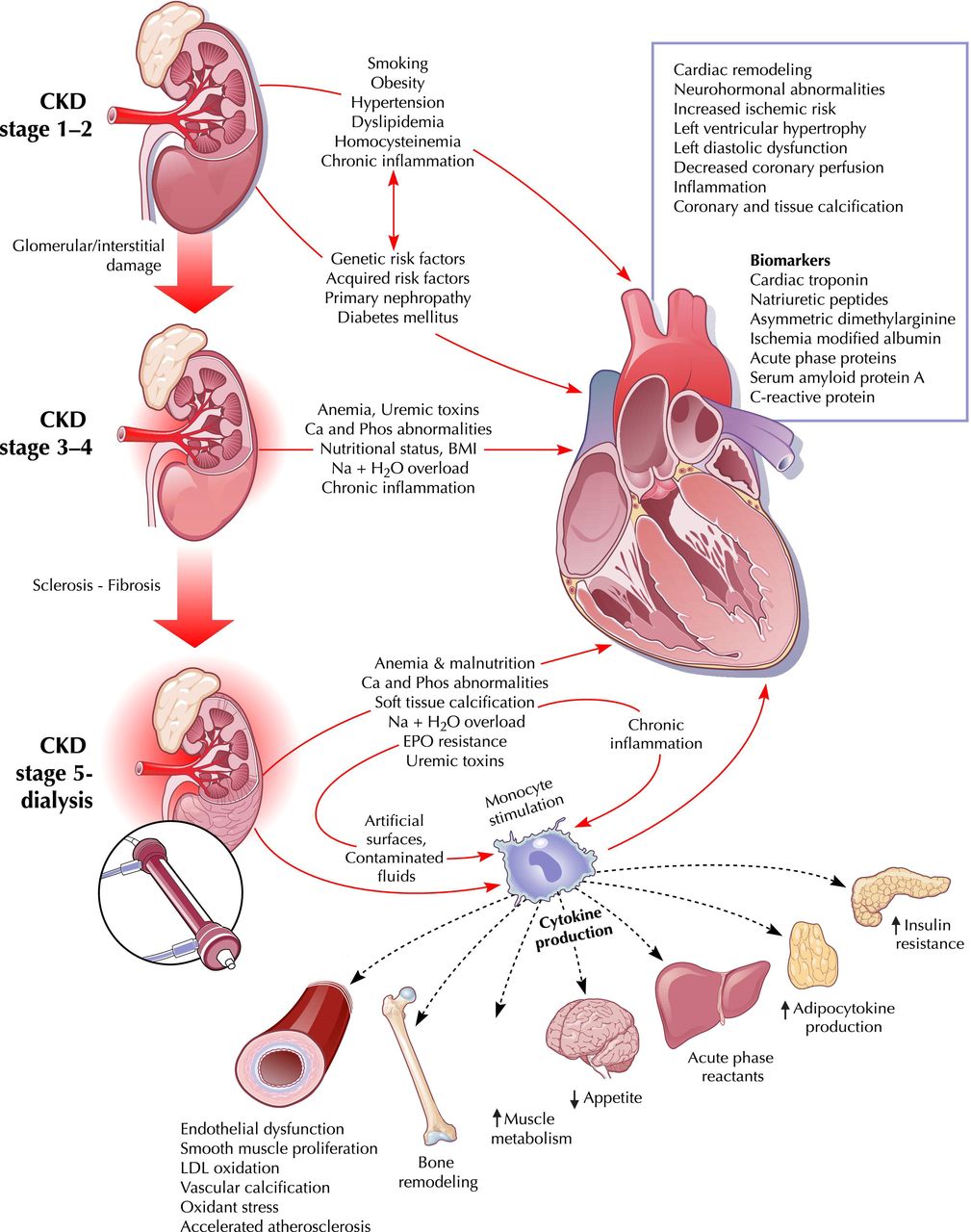
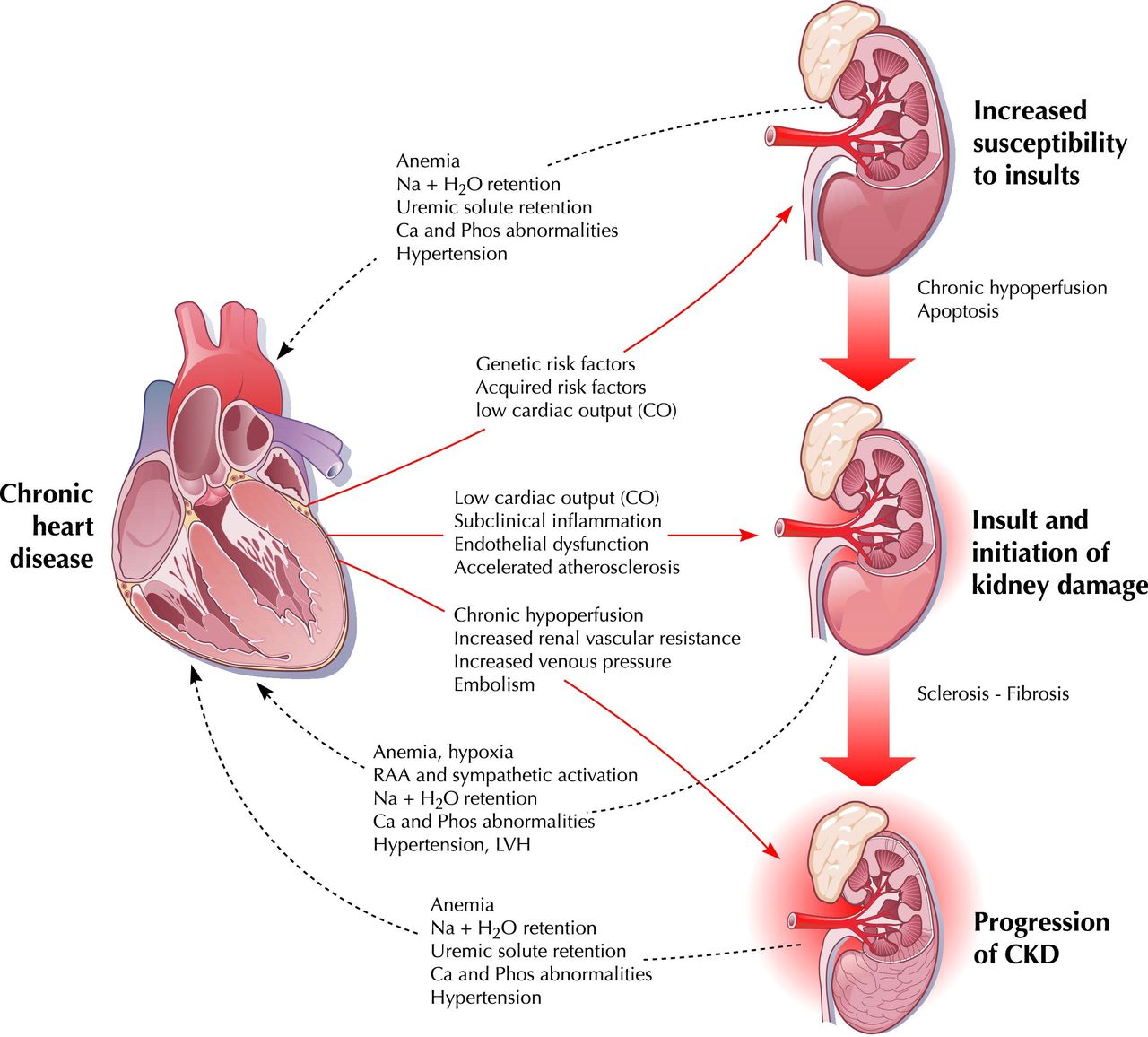
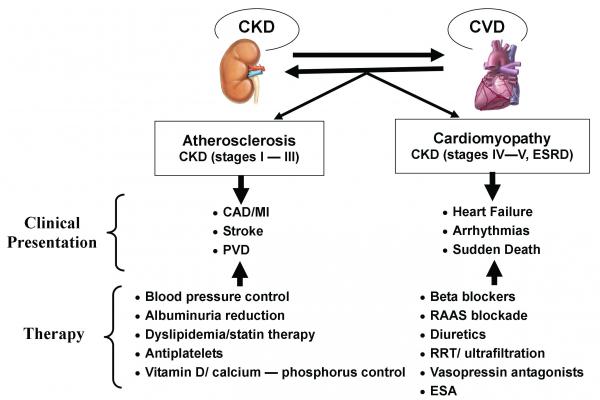
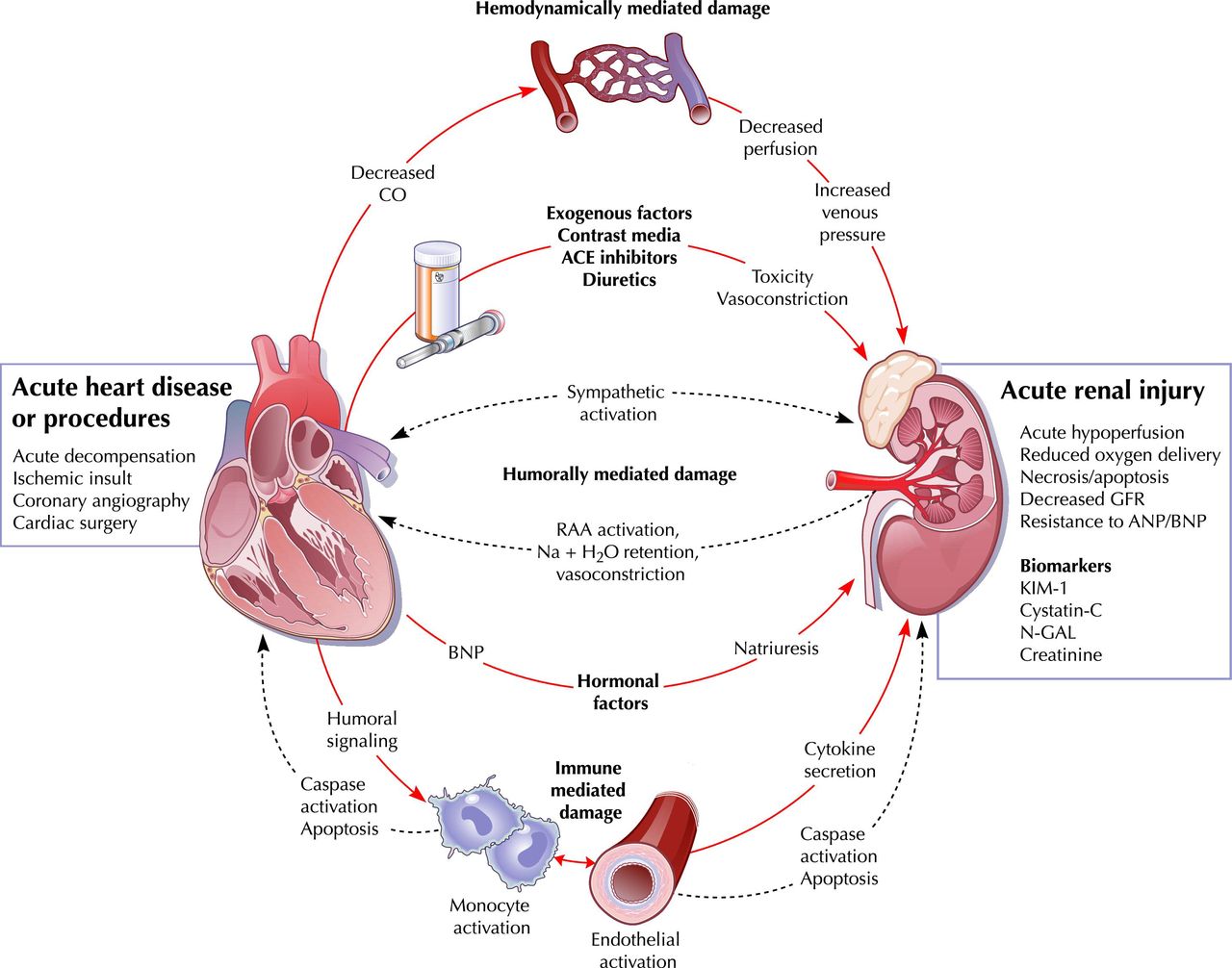
Cardiorenal syndrome (CRS) is an umbrella term used in the medical field that defines disorders of the heart and kidneys whereby “acute or chronic dysfunction in one organ may induce acute or chronic dysfunction of the other”.[1] The heart and the kidneys are involved in maintaining hemodynamic stability and organ perfusion through an intricate network. These two organs communicate with one another through a variety of pathways in an interdependent relationship. In a 2004 report from National Heart, Lung and Blood Institute, CRS was defined as a condition where treatment of congestive heart failure is limited by decline in kidney function.
The pathophysiology of CRS can be attributed to two broad categories of “hemodynamic factors” such as low cardiac output, elevation of both intra-abdominal and central venous pressures, and non-hemodynamic factors or “cardiorenal connectors” such as neurohormonal and inflammatory activation.[5] It was previously believed that low cardiac output in heart failure patients result in decreased blood flow to the kidneys which can lead to progressive deterioration of kidney function. As a result, diuresis of these patients will result in hypovolemia and pre-renal azotemia
progressive familial intrahepatic cholestasis
progressive familial intrahepatic cholestasis is a liver disease, which typically leads to liver failure. In people with PFIC, liver cells are less able to secrete bile which builds up in the liver causing liver damage.

Signs and symptoms of PFIC typically begin in infancy and are related to bile buildup and liver disease. Specifically, affected individuals experience severe itching, yellowing of the skin and whites of the eyes (jaundice), failure to gain weight and grow at the expected rate (failure to thrive), high blood pressure in the vein that supplies blood to the liver (portal hypertension), and an enlarged liver and spleen (hepatosplenomegaly).
There are three known types of PFIC: PFIC1, PFIC2, and PFIC3. The types are also sometimes described as shortages of particular proteins needed for normal liver function. Each type has a different genetic cause.
In addition to signs and symptoms related to liver disease, people with PFIC1 may have short stature, deafness, diarrhea, inflammation of the pancreas (pancreatitis), and low levels of fat-soluble vitamins (vitamins A, D, E, and K) in the blood. Affected individuals typically develop liver failure before adulthood.
The signs and symptoms of PFIC2 are typically related to liver disease only; however, these signs and symptoms tend to be more severe than those experienced by people with PFIC1. People with PFIC2 often develop liver failure within the first few years of life. Additionally, affected individuals are at increased risk of developing a type of liver cancer called hepatocellular carcinoma.
Most people with PFIC3 have signs and symptoms related to liver disease only. Signs and symptoms of PFIC3 usually do not appear until later in infancy or early childhood; rarely, people are diagnosed in early adulthood. Liver failure can occur in childhood or adulthood in people with PFIC3.
https://ghr.nlm.nih.gov/condition/progressive-familial-intrahepatic-cholestasis
Limb-girdle muscular dystrophies (LGMD)
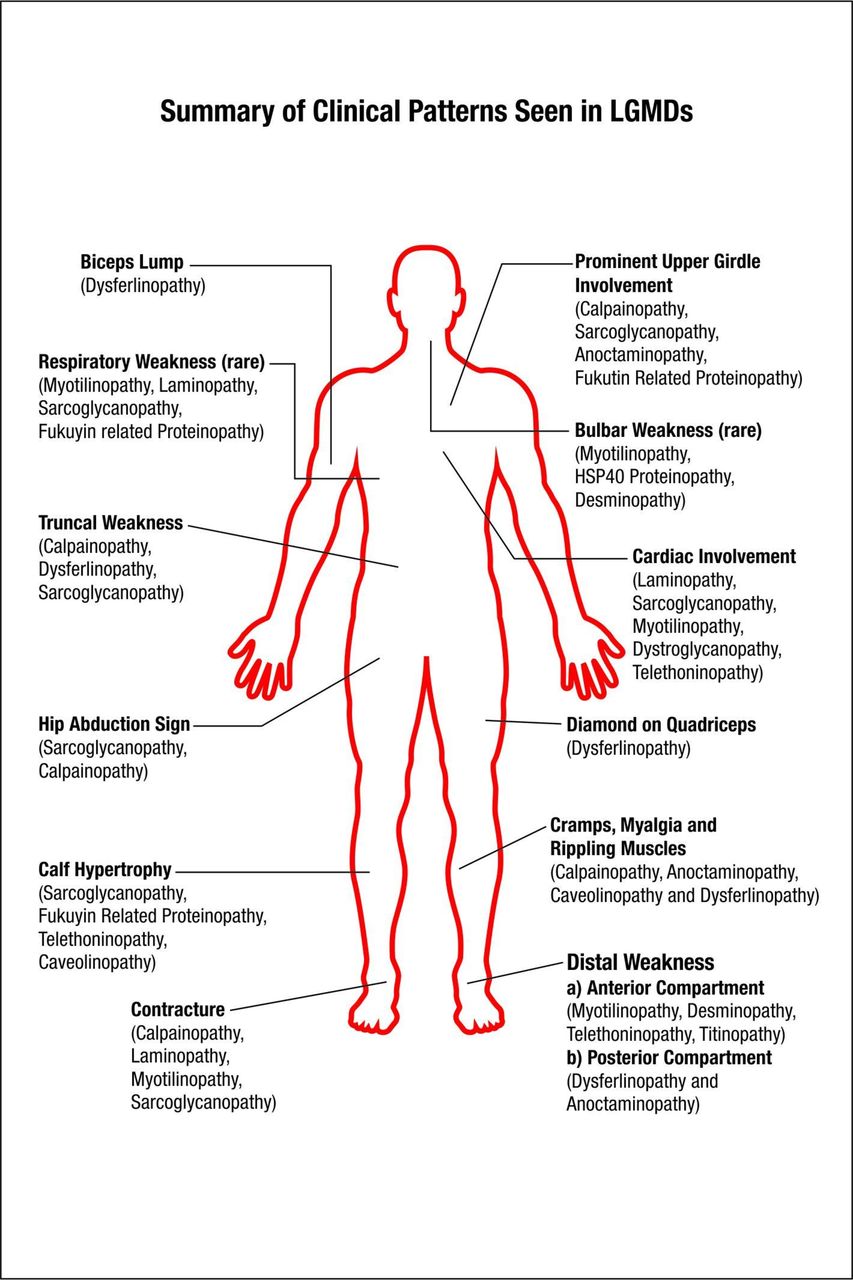
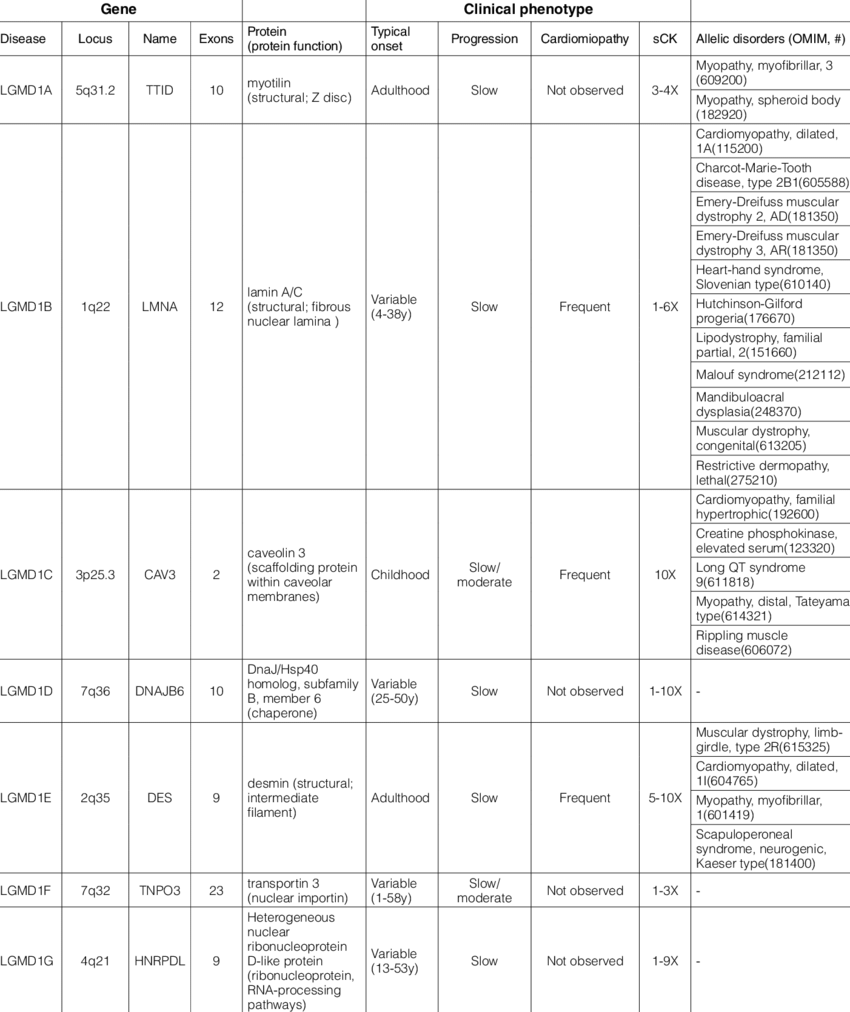
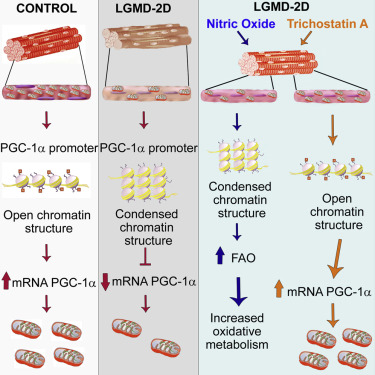
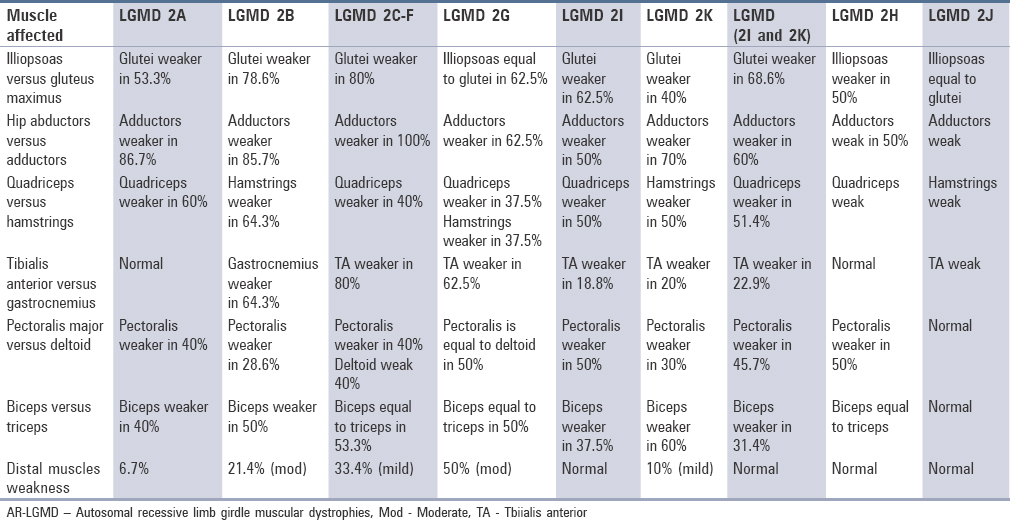
Limb-girdle muscular dystrophies (LGMD) are a group of rare progressive genetic disorders that are characterized by wasting (atrophy) and weakness of the voluntary muscles of the hip and shoulder areas (limb-girdle area). Muscle weakness and atrophy are progressive and may spread to affect other muscles of the body. Many different subtypes have been identified based upon abnormal changes (mutations) of certain genes. The age at onset, severity, and progression of symptoms of these subtypes may vary greatly from case to case, even among individuals in the same family.
https://rarediseases.org/rare-diseases/limb-girdle-muscular-dystrophies/

Extracorporeal membrane oxygenation (ECMO)
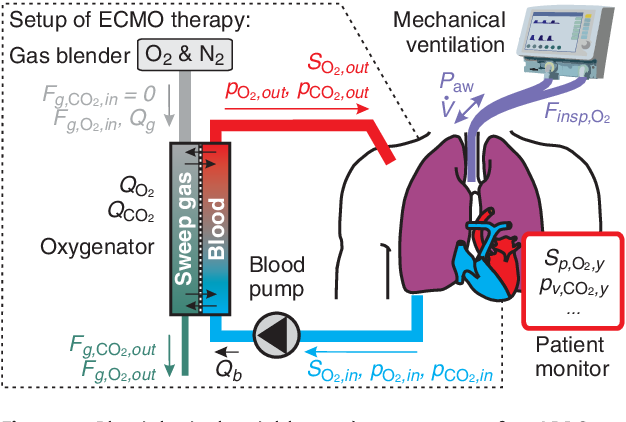
The ECMO machine replaces the function of the heart and lungs. For example, ECMO is used during life-threatening conditions such as severe lung damage from infection, or shock after a massive heart attack.
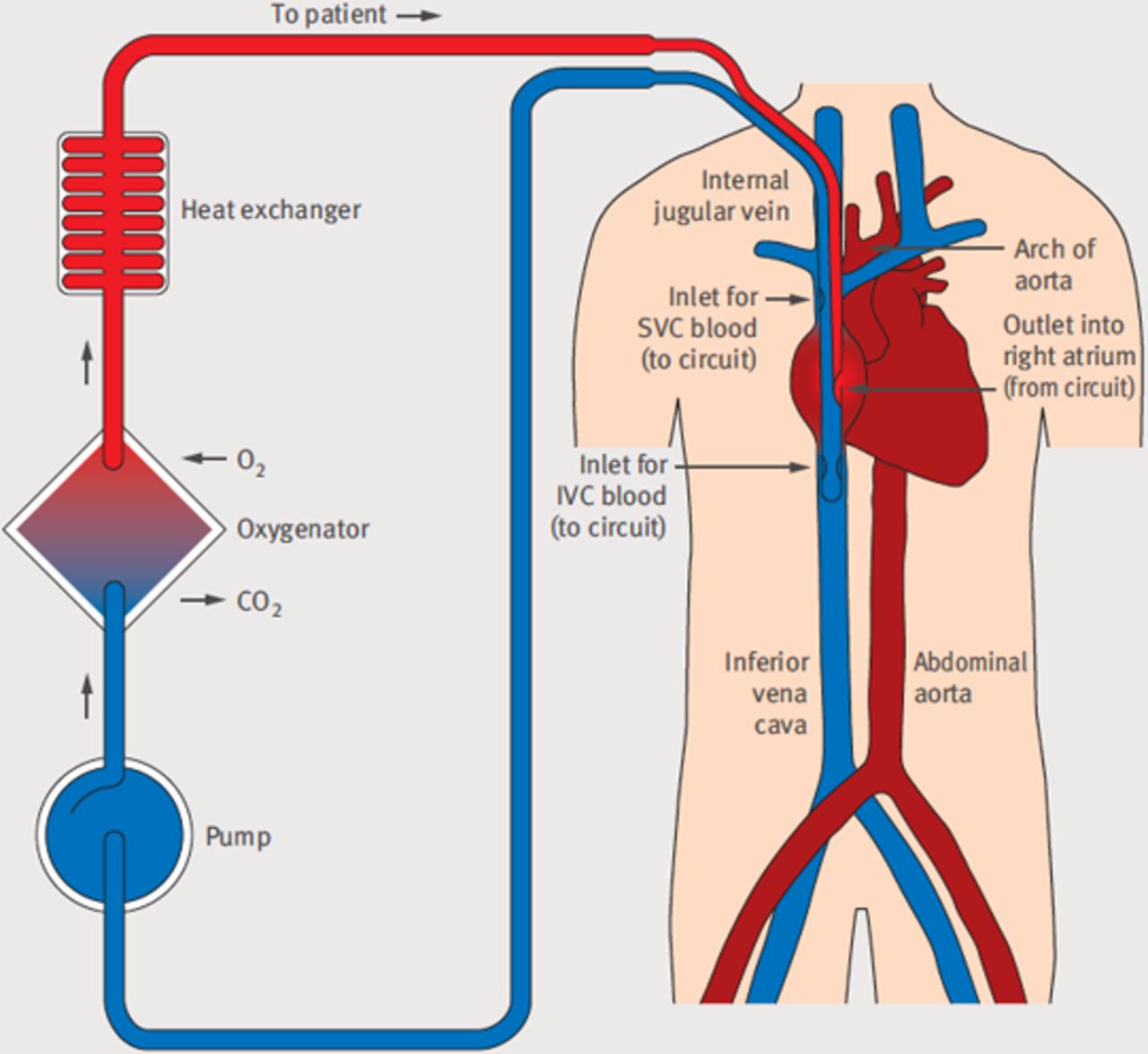
ECMO is used to help people whose:
■■ Lungs cannot provide enough oxygen to the body even when given extra oxygen
■■ Lungs cannot get rid of carbon dioxide even with help from a mechanical ventilator
■■ Heart cannot pump enough blood to the body
ECMO may also be used to support people with heart or lung dis- ease that cannot be cured while they wait for an organ transplant (e.g. new heart and/or lungs).
The ECMO machine pumps blood from the patient’s body to an artificial lung (oxygenator) that adds oxygen to it and removes carbon dioxide. Thus, it replaces the function of the person’s own lungs. The ECMO machine then sends the blood back to the patient via a pump with the same force as the heart, replacing its function. The ECMO machine is controlled by a person called a perfusionist, or a nurse or respiratory therapist with advanced training called an ECMO specialist. The perfusionist or ECMO specialist will adjust the settings on the machine to give the patient the amount of heart and lung support they need.
Click to access what-is-ecmo.pdf
Acetabuli protrusio
Protrusio acetabuli is a rare anatomic pattern of the hip in which the femoral head protrudes into the true pelvis.
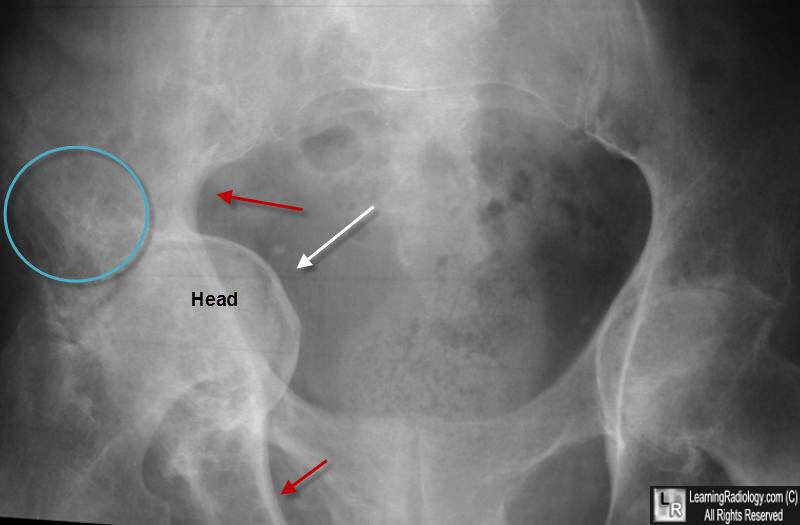
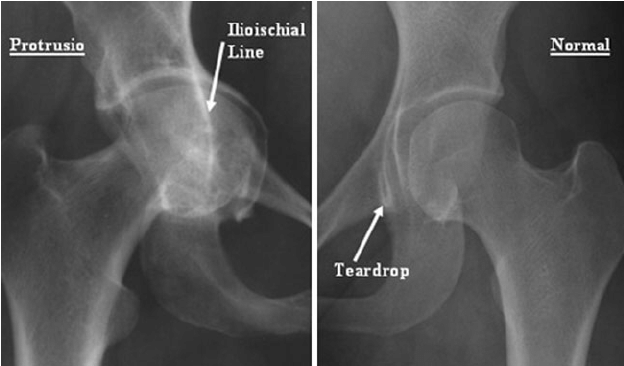
Protrusio acetabuli (arthrokatadysis or Otto pelvis) is a relatively rare condition associated with secondary osteoarthritis of the hip. Radiographically, protrusio acetabuli is present when the medial aspect of the femoral head projects medial to Kohler’s (ilioischial) line. This results in medialization of the center of rotation (COR) of the hip. Protrusio acetabuli is typically associated with metabolic bone disease (osteoporosis, osteomalacia, Paget’s disease) or inflammatory arthritis (RA or ankylosing spondylitis). Idiopathic acetabular protrusio can occur without the above associated factors however. Patients with protrusio acetabuli typically present with significant restriction of range of motion (ROM) of the hip due to femoral neck and trochanteric impingement in the deep acetabular socket and pain associated with secondary osteoarthritis (OA).
https://online.boneandjoint.org.uk/doi/abs/10.1302/1358-992X.97BSUPP_1.CCJR2014-031



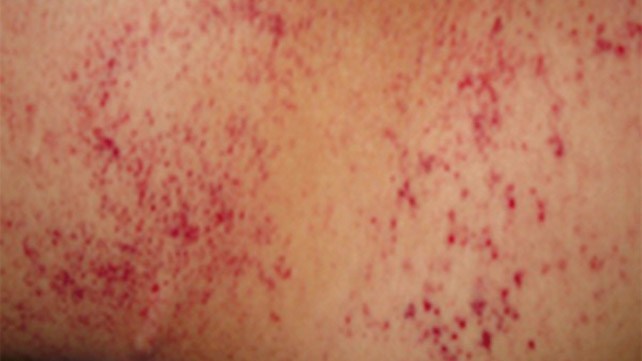
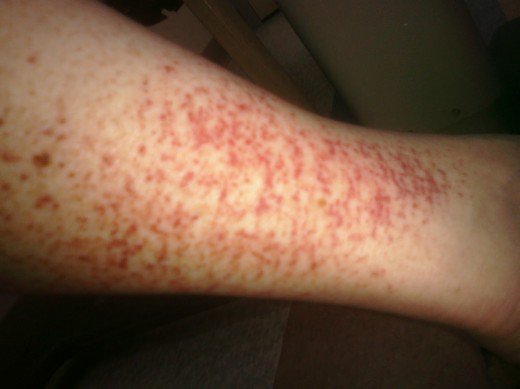
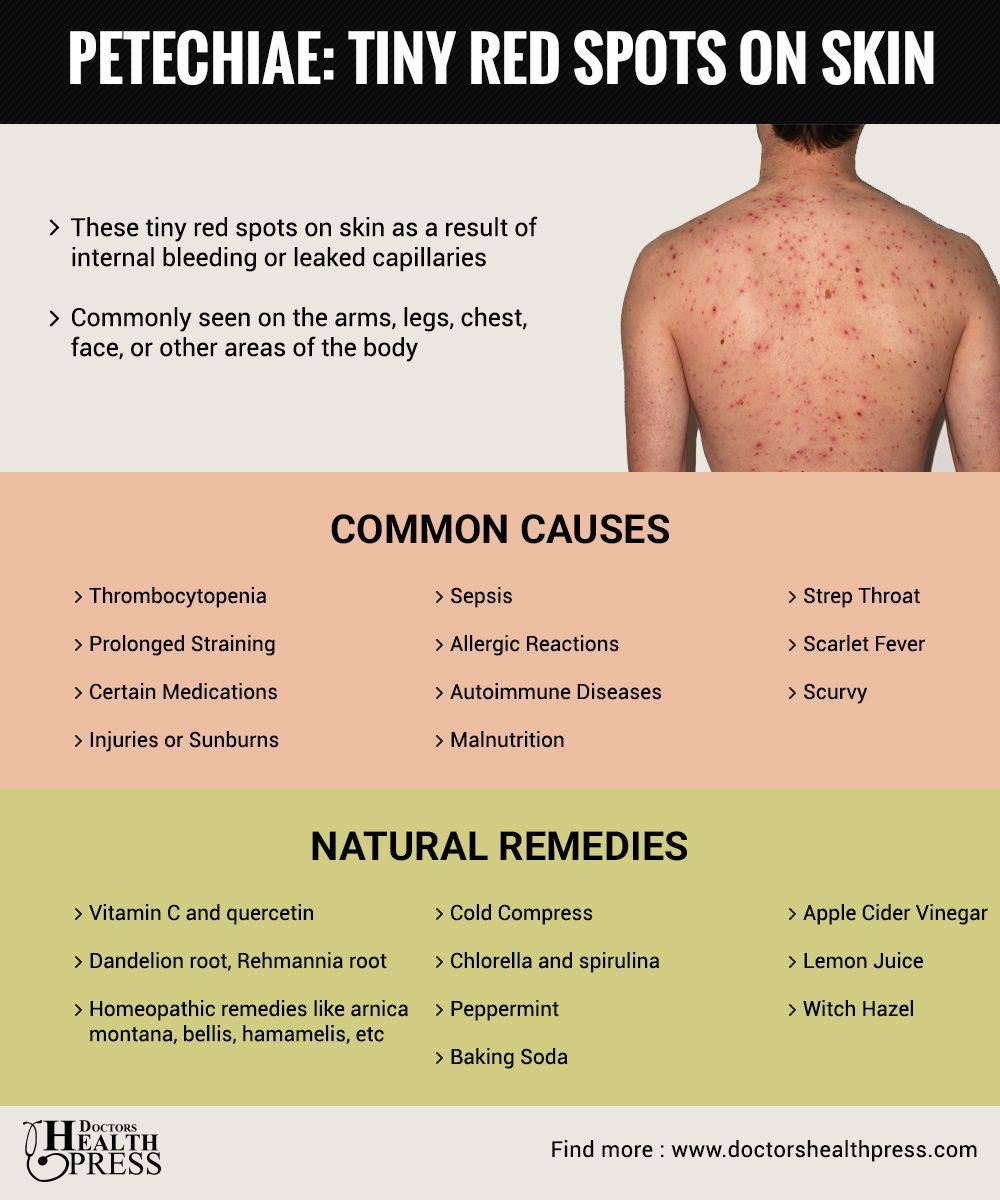
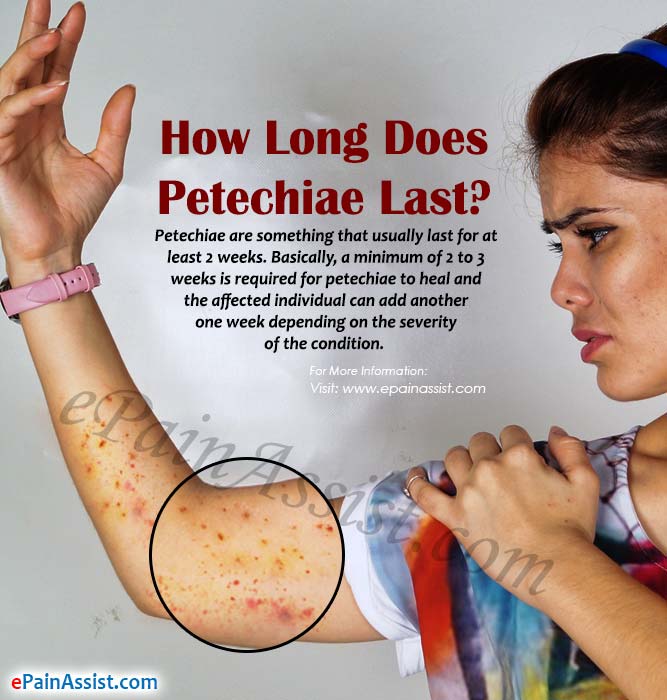
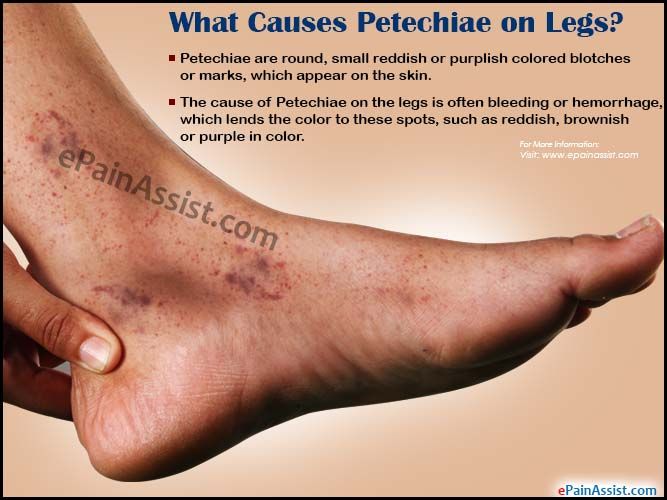
Petechiae
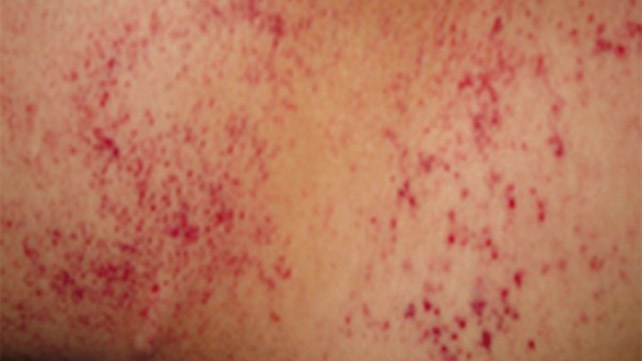
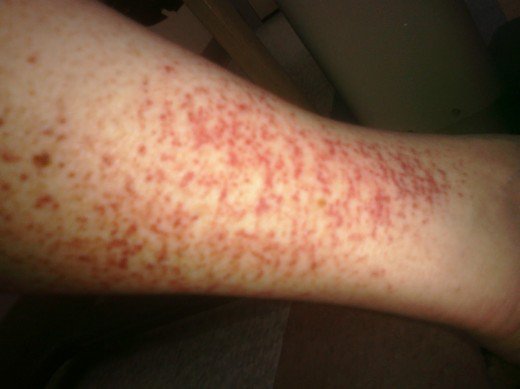
Petechiae are tiny, circular, non-raised patches that appear on the skin or in a mucous or serous membrane. They occur as the result of bleeding under the skin.
Causes
Petechiae occur when tiny blood vessels (capillaries) break open. When this happens, blood leaks into the skin.
Some of the conditions that may result in the appearance of petechiae include:
- local injury or trauma causing damage to the skin
- sunburn
- allergic reactions to insect bites
- various autoimmune diseases
- viral and bacterial infections
- a lower than normal blood platelet level
- medical treatments for cancer, such as radiation or chemotherapy
- leukemia or bone marrow problems that can cause a reduction in the number of platelets
- after violent vomiting or coughing — especially in newborns
- strenuous activity that may cause straining, such as lifting weights or giving birth
- sepsis
- scurvy
- vasculitis
- viral fevers, such as dengue, Ebola, and yellow fever, can inhibit blood clotting, causing bleeding under the skin
Certain medications are also commonly associated with the appearance of petechiae. Drugs that may cause petechiae as a side effect include:
- antibiotics
- antidepressants
- anti-seizure drugs
- blood thinners
- heart rhythm drugs
- nonsteroidal anti-inflammatory drugs (NSAIDs
- sedatives